คณะวิทยาศาสตร์

Development of a Nested PCR Assay to Avoid False Positive Detection of the Microsporidian Enterocytozoon hepatopenaei in Environmental Samples in Shrimp Farms
October 24, 2019
Transplantation of Macaca cynomolgus iPS – derived Hematopoietic Cells in NSG Immunodeficient Mice
October 24, 2019Accelerated Telomere Shortening in β – thalassemia/HbE Patients
Faculty of Science, Mahidol University
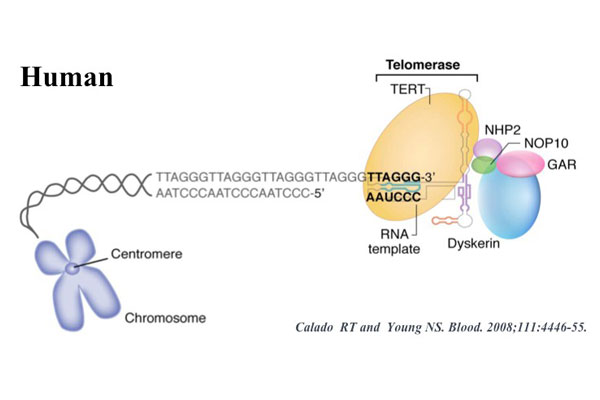
β-Thalassemia/HbE disease is caused by a defective β-globin synthesis that leads to accumulation of excess unbound α-globins, and consequently oxidative stress, ineffective erythropoiesis and chronic anemia. Cell replication and oxidative stress are factors contributing to erosion of telomeres responsible for maintaining genomic stability and cell replication capability. In this study, the rate of telomere shortening in β-thalassemia/HbE patients was compared to the rate of telomere shortening in normal individuals. Telomere length was determined from peripheral blood mononuclear cells of 43 β-thalassemia/HbE patients and 22 normal controls using Flow-FISH analysis. The telomere length was shown to be age-dependent in normal group (rs = 0.715, P = 0.002), whereas severity-dependent telomere shortening was observed in the patients. The telomere length of patients who had severe clinical symptoms (10.07 ± 2.15%) was shorter than that of patients who showed mild symptoms (15.59 ± 2.27%), moderate symptoms (14.50 ± 1.41%) and those in the normal group (14.75 ± 3.11%, P < 0.05). Additionally, reticulocyte count and oxidative stress were correlated with telomere length. This indicates that increased oxidative stress and markedly enhanced erythropoiesis in β-thalassemia/HbE patients leads to accelerated telomere erosion in clinically severe patients.

Dr. Porntip Chaichompoo
Faculty of Science, Mahidol University
porntip.cha@mahidol.ac.th